Klippel-Feilov sindrom: znakovi, fotografija, liječenje, prognoza
Godine 1912. dva neurologa francuskog podrijetla, Andre Feil i Maurice Klippel, detaljno su opisali urođenu deformaciju cervikotorakalne kralježnice koja se pojavila u djece. Svi bolesnici s ovom bolešću su djeca roditelja koji imaju krvnu vezu.
To ne čudi, budući da su u srednjem vijeku rođaci mogli postati zakonski supružnici. Malo kasnije, ta je patologija nazvana u čast znanstvenika. Zapravo, Klippel-Feil sindrom nije bolest. To je kongenitalna patologija koja može uzrokovati razvoj drugih bolesti kralježnice. 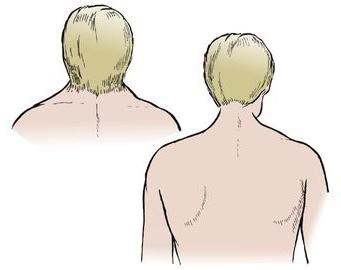
Značajke sindroma
Ovaj sindrom je prirođena patologija vratne kralježnice, koja smanjuje broj kralježaka i synostosis. Za njega su karakteristične vrlo česte manifestacije: opaženo je kod jednog novorođenčeta od 120 tisuća. Najtipičniji je skraćeni vrat. Većina slučajeva bolesti popraćena je drugim patologijama koje utječu na mišićno-koštani sustav i unutarnje organe. Dijagnoza ove bolesti leži u sposobnosti takvih uskih stručnjaka kao što su neurolog, genetičar, ortoped i još neki. Liječenje konzervativnog tipa može se sastojati od masaže, fizioterapije i vježbanja. Ako je slučaj ozbiljan, tada nije isključena potreba za kirurškim liječenjem, koji se sastoji od cervikalizacije.
Mora se napraviti razlika između genetskih defekata i češćeg tortikolisa ili sindroma lažnog kratkog vrata kod novorođenog djeteta, što je rezultat traume rođenja. Kada se novorođenče pomiče duž rodnog kanala, vratni kralješci se komprimiraju, što čini iluziju da je vrat kratak. Takve anomalije mogu se ispraviti, one obično nestaju bez traga tijekom prve godine života.
Uzroci Klippel-Feil sindroma
Ovaj sindrom je uključen u skupinu genetski uvjetovanih bolesti. Patološke promjene počinju se razvijati u prvim tjednima razvoja fetusa u maternici. Liječnici bilježe nekoliko glavnih razloga zbog kojih se sindrom manifestira. To uključuje oslabljenu segmentaciju i razvoj kralješnice, uglavnom na gornjoj razini cerviksa.
Što pomaže u određivanju kliničke slike?
Klinička slika bolesti može se odrediti:
- Neuspjeh lukova i tijela.
- Smanjenje broja kralježaka.
- Synostoze vratnih i prsnih kralješaka.

U rizičnu skupinu spadaju djeca koja imaju neuspješno nasljedstvo, a karakteriziraju:
- Genetski defekti u kromosomima. Dijete je poremećeno formiranjem diferencijacije rasta, koja je neophodna da bi se kostur u potpunosti razvio. Ovo kršenje utječe na razvoj prsnih i vratnih kralješaka.
- Autosomno dominantno nasljeđivanje. To znači sljedeće: ako jedan od roditelja ima bolest, tada će se bolesno dijete roditi s vjerojatnošću od 50-100 posto.
- Autosomno recesivni tip nasljeđivanja. U ovom slučaju, vjerojatnost se smanjuje i kreće se od nule do pedeset posto.
Kako bi se izbjeglo disfunkcionalno nasljeđivanje, roditelji bi trebali potražiti savjet od genetike prije zasnivanja djeteta.
Znakovi Klippel-Feil sindroma
Ovu bolest karakterizira klasična trijada simptoma:
- Mijenja granice rasta kose.
- Promatran prekratak vrat.
- Pokretljivost glave je ograničena.

Najčešće se kombinira s drugim bolestima. Oko trideset posto pacijenata s Klippel-Feil sindromom pati od skolioze, krutih oblika tortikolisa i visokog položaja lopatica, koje se nazivaju Sprengelova bolest. U nekim slučajevima, obilježene su abnormalnosti ruku, deformacije stopala, zubi. tamo asimetrija lica i dalekovidnost. Oko dvadeset pet posto pacijenata pati od urođene gluhoće.
Ovaj sindrom nije samo kozmetički defekt s jakom ozbiljnošću. Osim toga, pod njim leže ozbiljne neurološke komplikacije. Mogu se izraziti u razvoju oligofrenije, hidrocefalusa, epilepsije. Od ranog djetinjstva, pacijenti osjećaju mišićnu slabost udova, sinkinezu. U starijoj dobi klinička slika bolesti dopunjena je pojavom sekundarnih promjena u kralježnici.
Ako je dostupno simptomi sindroma Klippel-Feil, mora se ispitati.
Liječnički pregled
Sindrom se provjerava na temelju sadašnje trijade simptoma, instrumentalnog pregleda i fizikalnog pregleda. Posebnu ulogu u postavljanju dijagnoze ima proces proučavanja obiteljske povijesti bolesnika. Klippel-Feilov sindrom se može dijagnosticirati i povezane anomalije mogu se opisati koordiniranim radom uskih stručnjaka kao što su kardiolozi, neurolozi, ortopedi, genetika, pulmolozi.
Za procjenu prirode promjena kralježnice omogućuje se radiografija. Takvu bi studiju trebalo provesti u dvije projekcije, pri čemu je bočna projekcija više informativna. S obzirom na činjenicu da se glava nalazi neuobičajeno, sjena lubanje može se kombinirati s slikom kralježnice, koja, pak, ne dopušta otkrivanje detalja. Također se preporučuje snimanje dodatnih slika koje prikazuju maksimalnu fleksiju i proširenje vrata. Oni su ti koji omogućuju otkrivanje nestabilnosti ne akkretnih kralješaka. Što je još uključeno u dijagnozu Klippel-Feil sindroma? 
Radiografsko-torakalna radiografija kralježnicom omogućuje vam otkrivanje:
- Prisutnost deformiranih kralješaka.
- Prirast tijela kralješaka.
- Smanjite njihov broj.
- Zakrivljenost kralježnice.
- Nenormalan položaj oštrica.
Nakon što je dijagnoza potvrđena, liječnik propisuje dodatni postupak ultrazvuka unutarnjih organa. To vam omogućuje da identificirate anomalije u njihovoj prisutnosti. Ako je sindrom popraćen neurološkom patologijom, možda će biti potrebno provesti ultrazvuk krvožilnog ultrazvuka, MRI cervikalne regije, EEG i angiografiju. Savjetovanje s genetikom je obvezno. Na temelju rezultata liječnik može odrediti koja je vrsta bolesti nasljedna i koja je opasnost od pojave bolesti u budućim generacijama.
Oblici sindroma
Do danas je Klippel-Feilov sindrom (fotografije se može vidjeti u ovom članku) rijetka patologija. Simptomi bolesti dijagnosticiraju se kod jednog djeteta od sto dvadeset tisuća. Ova anomalija ima tri oblika: 
- Smanjuje se broj segmenata cervikalne regije, postupno rastu zajedno, što rezultira vizualnim skraćivanjem vrata. Ovaj oblik bolesti uzrokuje teško kretanje glave.
- Formirana je synostoza cervikalne kralježnice kao posljedica njezine fuzije s potiljnom kosti. S ovim oblikom, pacijent gubi sposobnost okretanja glave bez boli. Potiljak i vratni kralješci su jedna cjelina.
- Kombinacija prva dva tipa patologije je treći oblik.
Samo stručnjak s rezultatima potrebnih istraživanja može odrediti oblik sindroma.
Načela liječenja bolesti
Nažalost, moderni liječnici nemaju adekvatne metode za potpunu eliminaciju sindroma. Terapija uključuje samo prevenciju pojave sekundarnih deformiteta. Najčešće se primjenjuju konzervativne metode liječenja koje se temelje na masaži i vježbanju. Terapija lijekovima može se propisati kada je sindrom popraćen manifestacijom jakog bola, au tom slučaju, ako postoji kompresija korijena živaca. Najčešće se koriste lijekovi iz skupine analgetika, kao i lijekovi s protuupalnim učinkom. Ako je kompresija produljena, tada je indicirana kirurška intervencija. U tom slučaju, glavna svrha operacije je uklanjanje boli i djelomična korekcija vanjskih defekata.
Kirurško liječenje Klippel-Feil sindroma
Glavna indikacija za operaciju je stalna bol, koja uzrokuje kompresiju korijena živaca. Važno je napomenuti da se s godinama stanje bolesnika značajno pogoršava. Zato se operacija provodi bez odgađanja, odmah nakon dijagnoze sindroma. U cilju povećanja pokretljivosti vrata pacijenta, upotrebom tehnike kao što je cervikalizacija po metodi Bonola. Takva operacija uključuje uklanjanje četiri gornja rebra i periosta, što smanjuje pritisak na unutarnje organe. Provesti sličnu operaciju u nekoliko faza: prvo, kirurg uklanja rebra iz pacijenta s jedne strane, nakon čega slijedi prilično dugo razdoblje rehabilitacije i obnove tijela. Nakon toga se izvodi druga faza operacije, a rebra se skidaju s druge strane. 
Period rehabilitacije je vrlo dug i zahtijeva puno truda od strane pacijenta, njegove rodbine i liječnika. Pacijent koji je podvrgnut takvom kirurškom zahvatu morat će provesti nekoliko mjeseci u stacionarnom stanju. Međutim, nemojte napustiti operaciju. Moderna medicina može pružiti samo jedan učinkovit način liječenja, a to je upravo takva operacija. To će eliminirati vanjske nedostatke. Ako se, međutim, odbije liječenje, može doći do razvoja komplikacija Klippel-Feil sindroma.
Moguće komplikacije
Prvi je pogoršanje bolesti unutarnjih organa, a zatim se razvija jak bolni sindrom. Bol je posljedica narušavanja korijena živaca. To, usput, može dovesti do potpune imobilizacije.
Bolesti unutarnjih organa ugrožavaju patološke procese koji su nepovratni. Rezultat može biti prijevremena smrt pacijenta. 
pogled
Kakva je prognoza za Klippel-Feil sindrom?
Ako su operacija i razdoblje rehabilitacije uspješni, pacijent može računati na pun život. Kao rezultat operacije, pojavit će se vizualno produljenje vrata, što je povoljno s estetskog stajališta.
Preventivne mjere
Kao što smo već primijetili, ovaj sindrom je genetski određen, stoga ne postoji specifična prevencija ove bolesti. Ako je bilo slučajeva manifestacije slične patologije u obitelji, onda je obavezno posjetiti genetičara u fazi planiranja djeteta. Stručnjak će provesti potrebna istraživanja, procijeniti rizik od pojave sindroma Klippel-Feil u nerođenog djeteta. Posljedice se mogu izbjeći.